Immunotherapy Resistance and Inflammatory Memory
Inflammatory Memory as an Adaptive Response Co-opted by Cancer
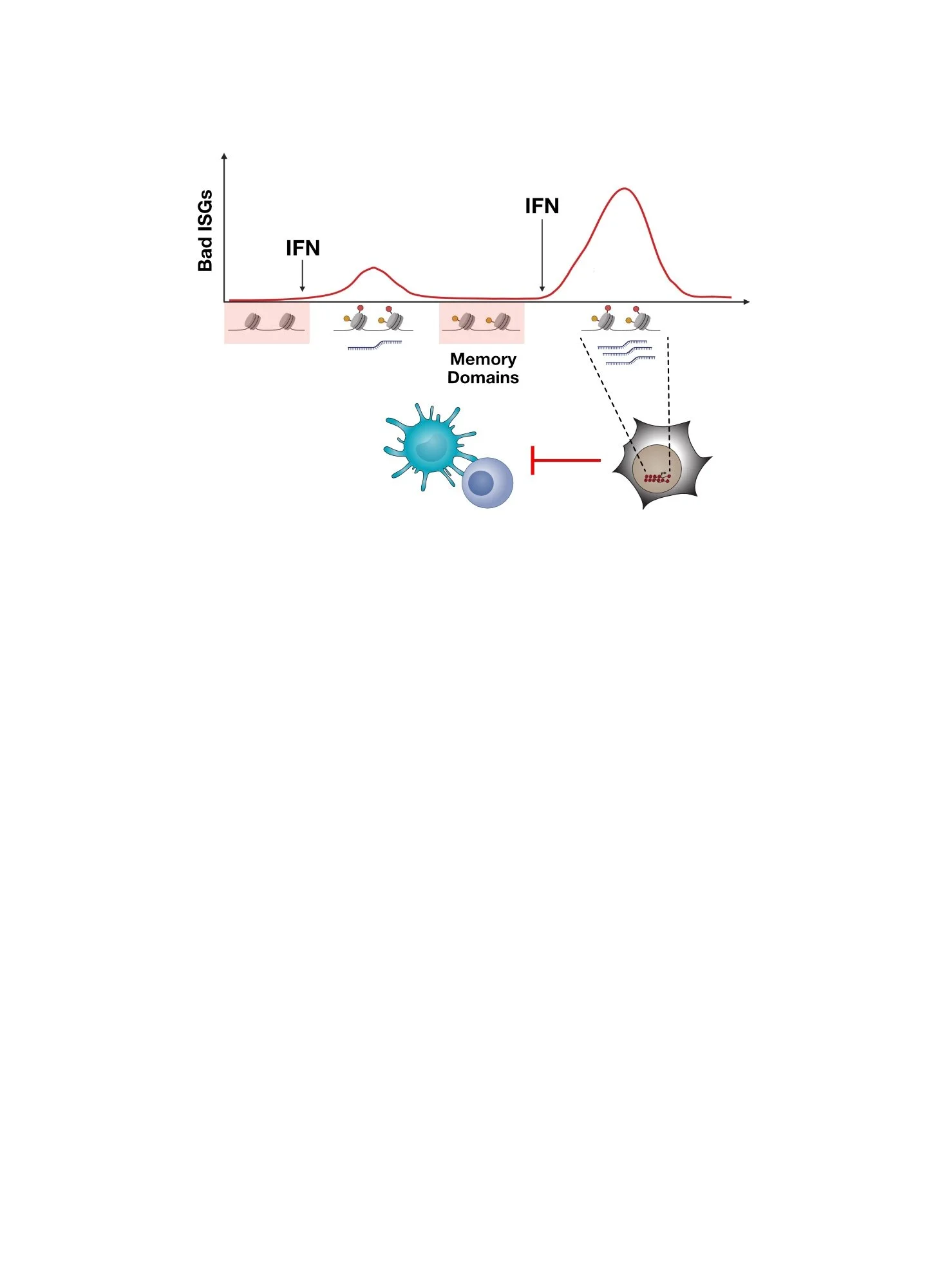
Inflammatory memory describes the process whereby cells “remember” previous inflammatory stimuli in order to alter subsequent responses to the same or similar inflammatory stimuli. But why would cells want to do this? In situations of chronic or recurring inflammation (e.g., chronic virus infection, autoimmunity), tissues may need to adapt to the effects of chronic stimulation in order to minimize ensuing detrimental effects (immune-mediated pathology). This adaptive response may improve tissue repair or down-modulate immune cells that can cause tissue damage from runaway inflammation. One mechanism for the cellular encoding of inflammatory memory involves epigenetic reprogramming. The extent to which inflammatory memory or its dysfunction contributes to human diseases is an intense area of investigation. Could inflammatory memory play a role in cancer?
Epigenetically encoded inflammatory memory in health and disease. Chronic inflammation can lead to adaptive responses in various cell types, enabling tissues and host to better avoid potential detrimental effects of persistent inflammation. In diseases such as cancer, inflammatory memory may be co-opted.
Immunotherapy Resistant Cancer Cells Acquire Epigenetic Features of Inflammatory Memory
In cancer, we speculate that inflammatory memory might be co-opted to enable cancer cells to suppress the immune response against it, like normal tissues might do to prevent immune-mediated pathology. In this regard, we have discovered that chronic type II interferon (IFNG), which is often produced in the tumor by immune cells, can license cancer cells to acquire epigenetic features of inflammatory memory. This is evidenced by acquisition of chromatin accessibility that persists even when interferon (IFN) signaling is prevented by genetic means (e.g., STAT1 knockout). These epigenetic changes are acquired by a subset of IFN-stimulated genes (ISGs) in both mouse and human tumors, often augmenting their expression. In mice, this is accompanied by the acquisition of immunotherapy resistance, which includes immunotherapies like immune checkpoint blockade (ICB). In humans, this is associated with tumors with low CD8 T cell activity, a surrogate for poor ICB response. Among the ISGs linked to inflammatory memory include OAS1.
Epigenetic features of inflammatory memory acquired by immune checkpoint blockade (ICB) resistant tumors in mice (top) and human tumors with low CD8 T cell activity (bottom). Shown is chromatin accessibility around the OAS1 gene. In mouse tumors, IFN signaling was prevented by STAT1 KO, demonstrating persistent chromatin accessibility consistent with inflammatory memory.
Resistant Cancer Cells Use Inflammatory Memory-Related Genes to Amplify IFN-I Signaling
What is OAS1 , what does it have to do with inflammatory memory, and why might it contribute to immunotherapy resistance? OAS is a double-stranded RNA (dsRNA) sensor that has been shown to amplify type I IFN (IFN-I) signaling, possibly in coordination with other dsRNA sensors like RIG-I. Indeed, compared to their sensitive counterparts, resistant cancer cells with epigenetic features of inflammatory memory show greater IFN-I production and ISG expression when stimulated through agents that activate IFN signaling (like dsRNA ligands). This enhanced response requires OAS1. Thus, OAS1 fits the description of an inflammatory memory gene that contributes to the altered responses that cells with inflammatory memory acquire. But what about immunotherapy and specifically ICB resistance?
OAS1 is an inflammatory memory gene that amplifies IFN-I signaling and the expression of a subset of ISGs in immunotherapy resistant cancer cells. Shown is IFN-I production and ISG expression after stimulation with polyI:C, an agent that activates IFN signaling (left), and expression of ISGs in resistant melanoma and breast cancer cells with and without OAS1 knockout (right).
Amplified IFN-I Signaling Enables Cancer Cells to Sustain CD8 T Cell Exhaustion and Resistance
Do resistant cancer cells having acquired inflammatory memory and an enhanced IFN response rely on amplified IFN-I signaling to drive immunotherapy resistance? If so, what aspects of an anti-tumor immune response are impacted? To address these questions, we prevented IFN-I signaling in ICB-resistant cancer cells and then assessed whether features of immune cell function and the efficacy of ICB agents like anti-PD1 were improved. We found that in the tumor, CD8 T cells show evidence for greater interaction with certain subtypes of dendritic cells that typically support effective activation of T cells. Accordingly, these changes were accompanied by a greater proportion of CD8 T cells that appear effector-like and less exhausted (dysfunctional) after treatment with anti-PD1. Importantly, anti-PD1 response was also improved.
Preventing IFN-I signaling in ICB-resistant cancer cells improves predicted interactions between CD8 T cells and dendritic cells and increases the proportion of non-exhausted effector-like CD8 T cells in the tumor after anti-PD1 treatment. Shown are single-cell data for dendritic cells and CD8 T cell subtypes in the tumor (top). These immune changes are associated with restored response to anti-PD1 (bottom).
Targeting Inflammatory Memory Features and Using Drugs that Block IFN and Inflammatory Signaling to Restore Response
Besides driving CD8 T cell exhaustion and immunotherapy resistance, the amplified IFN-I signaling also serves to maintain the epigenetic features of inflammatory memory — a feed-forward loop. Deleting the transcription factor STAT1 (from IFN signaling) and IRF3 (from dsRNA sensor signaling) can revert many of the epigenetic features of inflammatory memory acquired by resistant cancer cells, thus breaking the loop.
Together, our findings argue for the importance of persistent IFN signaling in initiating resistance through inflammatory memory and maintaining the inflammatory memory state. Given this, we have been interested in blocking chronic IFN and inflammatory signaling with drugs like JAK inhibitors to improve immunotherapy efficacy. Indeed, in pre-clinical models JAK inhibitors given after ICB can improve ICB response.
Amplified IFN-I signaling by immunotherapy resistant cancer cells maintains the epigenetic features of inflammatory memory through STAT1 and IRF3. Shown are chromatin accessibility of inflammatory memory domains and for OAS1 (top). Blocking chronic IFN signaling using JAK inhibitors can improve anti-PD1 efficacy of otherwise ICB-resistant tumors (bottom).
Interested in learning more?
Visit our publications page for the full references to our work.
Qiu and Xu et al., Nature Cancer, 2023.
Here Are Some of the People That Led The Studies and Made It Happen!
Jingya Qiu, former grad student, current post-doc
Bihui Melidosian (Xu), former grad student








